
PNU 리서치
- 메인으로 이동
- 연구/산학
- PNU 리서치

비만으로 인해 지방조직이 손상되면, 이 과정에서 죽은 지방세포가 간 건강까지 악화시킬 수 있다는 연구 결과가 나왔다.
제약학과 황성환 교수팀은 비만으로 손상돼 죽은 지방세포가 염증성 면역세포인 S100A8 양성 대식세포를 간으로 끌어들여 지방 축적을 늘리고, 그 결과 대사이상 지방간질환(MASLD)을 악화시킨다는 분자 기전을 새롭게 밝혀냈다.
대사이상 지방간질환은 간에 지방이 과도하게 쌓이는 질환으로, 전 세계 성인 약 30%에서 나타나는 대표적인 대사질환이다. 비만이 주요 위험인자로 꼽혀 왔지만, 비만 상태에서 죽은 지방세포가 어떤 과정을 거쳐 간세포에 지방을 더 쌓이게 만드는지는 명확히 알려지지 않았다.
이번 연구에서는 비만 상태의 지방세포가 사멸하면서 유리지방산(FFA)과 세포외소포체(EV)를 방출하고, 이 신호가 간 안에 S100A8 양성 대식세포를 축적시킨다는 사실을 확인했다.
대식세포는 몸속에서 이물질을 제거하고 염증 반응을 조절하는 면역세포다. 이 가운데 S100A8 양성 대식세포는 염증 반응과 밀접한 S100A8 단백질을 많이 만드는 특정 대식세포 집단이다. 이 대식세포들이 간으로 이동하면, 원래 간세포의 지방 흡수를 억제하는 단백질인 CCN3의 발현이 줄어든다. CCN3는 세포 성장과 대사 조절에 관여하는 단백질로, 간세포 표면에서 지방산을 받아들이는 통로 역할을 하는 CD36의 발현을 억제하는 기능이 있다.
하지만 S100A8 양성 대식세포가 늘어나면 CCN3가 감소하고, 그 결과 CD36 발현이 증가해 간세포가 지방산을 더 많이 흡수하게 된다. 결국 간 안에 지방이 과도하게 축적되면서 지방간질환이 더 심해지는 것이다.
연구팀은 이런 과정을 규명하기 위해 독창적인 유전자 변형 동물모델을 활용했다. 대식세포에서만 S100A8 유전자가 없도록 만든 마우스와 지방세포가 죽지 않도록 항세포사멸 유전자 BCL2를 과발현시킨 마우스를 구축해 지방세포 사멸과 간 염증, 지방 축적의 연관성을 추적했다.
또 단일세포 전사체 분석(scRNA-Seq)을 통해 지방세포 사멸 신호가 간 내 S100A8 양성 대식세포를 증가시킨다는 점도 확인했다. 단일세포 전사체 분석은 세포 하나하나의 유전자 발현 상태를 정밀하게 분석해 어떤 세포가 늘고 줄었는지, 어떤 기능을 하는지 파악하는 기술이다.
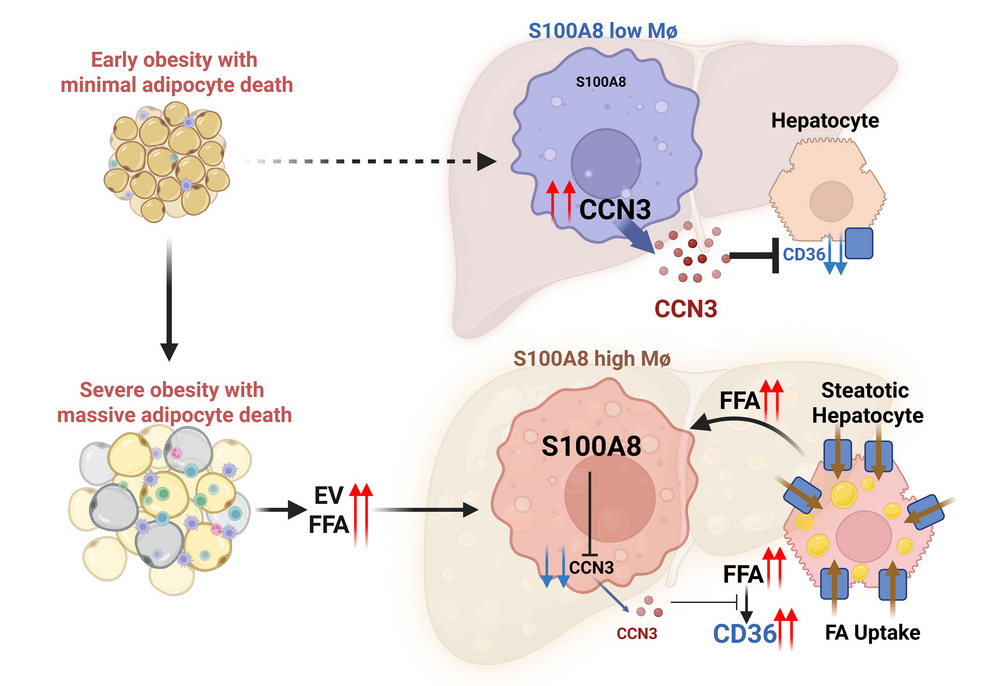
【S100A8 양성 대식세포에 의한 간세포의 지방축적 기전】
연구의 핵심은 지방간질환을 간 자체의 문제만으로 보지 않고, 지방조직과 간 사이의 상호작용이라는 관점에서 풀어냈다는 데 있다. 연구팀은 지방세포 사멸 → 간의 S100A8 양성 대식세포 증가 → CCN3 감소 → CD36 증가 → 간세포 지방 축적이라는 연쇄 경로를 제시하며, 비만이 지방간을 악화시키는 새로운 장기 간 상호작용 기전을 설명했다.
이번 연구는 미국 국립보건원(NIH) 산하 국립 알코올남용·알코올중독연구소(NIAAA) Bin Gao 박사 연구팀과의 국제 공동연구로 진행됐다. NIAAA 측이 동물모델 구축을, 부산대 연구팀이 분자 기전 분석을 맡아 융합 연구 성과를 냈다. 과학기술정보통신부 우수신진연구사업 지원을 받았으며, 제약학과 황성환 교수가 교신저자, 김연수 석박사통합과정생이 제1저자로 수행했다.
해당 연구 결과는 국제학술지 『Journal of Clinical Investigation』 2025년 11월 3일자에 게재됐다.
- 논문 제목: Adipocyte death promotes hepatic infiltration of S100A8+ macrophages and steatotic liver disease progression in mice (지방세포 사멸이 마우스에서 S100A8 양성 대식세포의 간 내 침윤과 지방간질환의 진행을 촉진한다)
- 논문 링크: https://www.jci.org/articles/view/190635
황성환 교수는 “이번 연구는 지방간질환의 원인을 간 자체가 아닌, 비만으로 손상된 지방조직과 간 사이의 상호작용에서 규명했다는 점에서 의미가 크다”며 “S100A8 양성 대식세포의 병인적 역할을 제시한 중요한 성과”라고 말했다.
연구팀은 이번 성과가 향후 지방세포 사멸, S100A8 양성 대식세포, CD36 매개 지방 흡수 경로를 겨냥한 새로운 지방간질환 치료제 개발에 중요한 단서를 제공할 것으로 기대하고 있다.
* 상단 사진: 연구팀. 왼쪽부터 김연수 석박사통합과정생, 박윤서 석박사통합과정생, 황성환 교수, 정준영 석사과정생, 조예은 석박사통합과정생.
[Abstract]
Both adipocytes and hepatocytes have the capacity to store fat, but the factor(s) that determine fat distribution between these cell types remain unknown. In mice fed a high-fat diet, fat initially accumulates predominantly in adipocytes, while hepatic fat accumulation mainly emerges after the onset of epididymal adipocyte death that results in elevated free fatty acids to promote lipid accumulation in hepatocytes. However, it remains unclear whether other signals after adipocyte death are required to direct and/or promote hepatocytes to store fat and subsequently trigger metabolic dysfunction-associated steatotic liver disease (MASLD, formerly known as nonalcoholic fatty liver disease). Using genetically modified mouse models combined with bulk and single-cell RNA-Seq analysis, we demonstrated that visceral adipocyte death induced an accumulation of S100A8+ macrophages in the liver, which was partially induced by fatty acids and apoptotic adipocyte-derived extracellular vesicles. Macrophage-specific deletion of the S100a8 gene reduced hepatic fat accumulation and MASLD severity in mice. Mechanistically, S100A8+ macrophages suppressed cellular communication network factor 3 (CCN3), a negative regulator of CD36, thereby enhancing CD36 expression in hepatocytes. In conclusion, adipocyte death promotes hepatic infiltration of S100A8+ macrophages, which drive hepatocyte lipid storage and subsequently promote MASLD progression through CD36 upregulation, partially mediated by CCN3 suppression.
- Authors (Pusan National University)
· First author: Yeonsoo Kim (College of Pharmacy)
· Corresponding author: Seonghwan Hwang (College of Pharmacy)
- Title of original paper: Adipocyte death promotes hepatic infiltration of S100A8+ macrophages and steatotic liver disease progression in mice
- Journal: Journal of Clinical Investigation
- Web link: https://www.jci.org/articles/view/190635
- Contact e-mail: shhwang@pusan.ac.kr